In collaboration with Karsten Jacobsen and S½ren Frederiksen in Denmark, we've
proposed a new method for estimating one source of prediction errors
for these models. We've found, for applications in both biology and physics,
that the parameters in these multiparameter models are sloppy -- varying in
concert by many orders of magnitude without making the fits
significantly worse! When we plug in these wildly varying almost-as-good
parameters into our models, we get a range of values for predicted
properties, which we can use to estimate "sloppy model" error estimates.
S½ren's thesis was on the interaction forces between atoms of the
element Molybdenum. He fit the many parameters of their force model to
training data generated by quantum calculations. These quantum
calculations have errors that for this purpose were negligable:
the errors in S½ren's prediction using the force model were systematic
errors (due to inadequacies in the model) rather than statistical errors
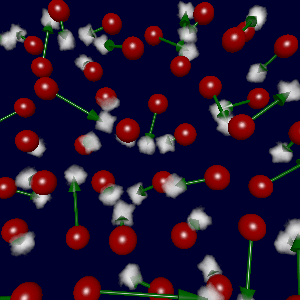
(due to errors in the fitted data). The figure at left shows the true
forces (green arrows) and forces generated using S½ren's potential (red),
with a sampling of parameters as described below.
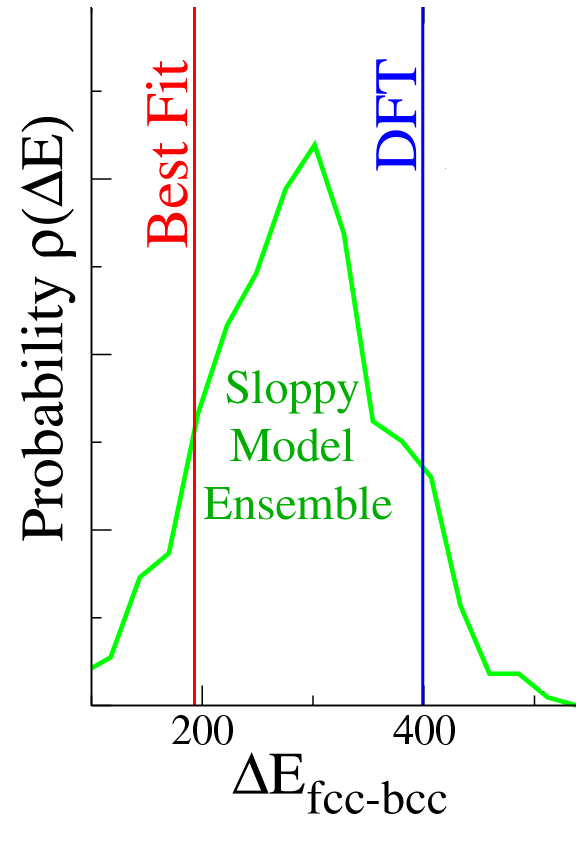
Can we get any idea for how big these systematic errors might be? For example, S½ren's best fit value for the energy difference between the fcc and bcc crystal structures (red line) was off from the true value (DFT, blue line) by more than a factor of two, while his predicted values for various elastic constants were often within one or two percent of the quantum calculations. (How accurate an old interatomic potential will be when used for a new purpose is called transferability.)
We suggested that S½ren try looking at the whole range of values for his
measured properties, allowing the parameters to vary away from the best
fit ones. Clearly fits that deviate from the best fit less than the
error in the best fit are also reasonable choices!
How do we sample these fluctuations? We do statistical mechanics
in model space, sampling them at a temperature
T0 set to make the fluctuations from the best fit
equal to the deviation of the best fit from the quantum calculation.
You see in the figure that the range of predictions at T0
(green curve) overlaps the true answer (vertical blue line).
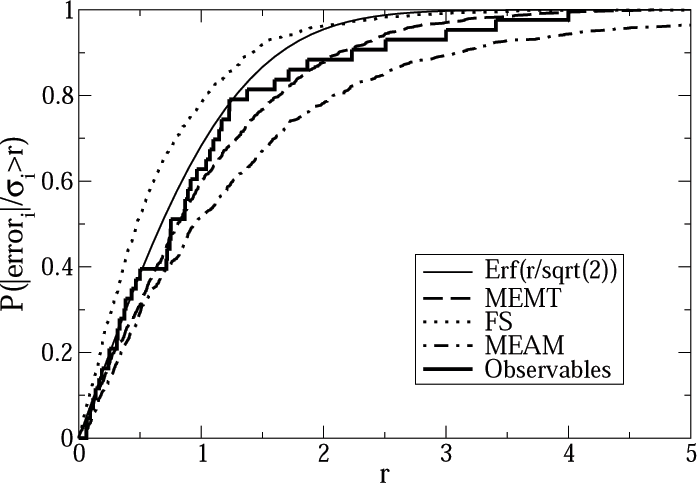
Using these alternative fits, S½ren generated alternative predictions to various materials properties of interest in molybdenum. He compared the the range of predictions (the `sloppy model' error predictions) to the actual error, found by directly calculating the predicted property using the more expensive quantum calculations and subtracting the best fit. The error estimates and the actual errors varied over a large range. The actual errors, though, were rather well predicted by the range of alternative predictions! We tested this by dividing the actual errors by the predicted errors: a perfect error prediction would give a Gaussian or normal distribution for this ratio. At right you see a plot of the integrated probability that this ratio is less than r: the Gaussian is shown by the solid line. The jagged curve gives the ratios for all the quantities S½ren calculated, using three different potentials: S½ren's MEMT potential, the classic Finnis-Sinclair potential, and a rival modern potential called MEAM. (The smooth curves represent predictions to force data for the three potentials.)
The sloppy model error was a good estimate, for Molybdenum, of the entire
systematic error! It isn't perfect: in the tail of the distribution:
S½ren finds predictions for which the sloppy model error is perhaps only
half of the entire systematic error. But now we can estimate, without
wisdom and years of experience, at least one source of error in these
calculations...
![]() James P. Sethna,
sethna@lassp.cornell.edu
James P. Sethna,
sethna@lassp.cornell.edu
 Statistical Mechanics: Entropy, Order Parameters, and Complexity,
now available at
Oxford University Press
(USA,
Europe).
Statistical Mechanics: Entropy, Order Parameters, and Complexity,
now available at
Oxford University Press
(USA,
Europe).